
In December, I created a blog post, "Decoding the Human Genome: Empowering Cancer Treatment Research with Oxford Nanopore MinION, AWS HealthOmics & AWS Bedrock," exploring six critical domains: Biology, Genetics, Diseases, and Whole Human Genome Sequencing using Oxford Nanopore MinION, AWS HealthOmics, and AWS Bedrock. Improving Pediatric Healthcare has always resonated with me, and I wanted to provide an example that personalized the capabilities of Genomics Secondary Analysis for precision medicine approaches.
The content I have put together continues on the previously mentioned blog post, starting with uploading the fastq.gz sequenced reference and patient human genome files, which are processed into VCF files from the Chromosomes Analysis. These end chromosome VCF output files are analyzed further to identify actionable mutations and apply targeted therapies that specifically inhibit these mutations. This particular example is for Pediatric Medulloblastoma, which is the most common malignant brain tumor in children, constituting nearly 20 percent of all pediatric brain tumors. In my next post, I plan to utilize AWS HealthOmics to demonstrate the efficiency benefits of performing similar Genomics Secondary Analysis Workflows.
Main Sections
Section 1 - Introduction
Section 2 - Genomics Sequencing Overview
Section 3 - Upload fastq.gz Sequence Files Into AWS S3 Bucket
Section 4 - Creating BWAMem, SAMTools, and BCFTools Docker Images and Upload to AWS S3 Bucket
Section 5 - Pushing BWAMem, SAMTools, and BCFTools Docker Images to AWS ECR Repositories
Section 6 - Creating the AWS Fargate Compute Environment
Section 7 - Creating the AWS Batch Queue Utilizing AWS Fargate Compute Environment
Section 8 - Creating the AWS Batch Job Definitions
Section 9 - Creating the AWS Step Functions State Machine
Section 10 - AWS Step Functions State Machine Workflow
Section 11 - AWS Step Functions State Machine Workflow Start-Execution and Output to AWS S3
Section 12 - Leveraging Genomic Data for Precision Treatment of Pediatric Medulloblastoma Example
Section 13 - A Simplified Path with AWS HealthOmics
Section 1 - Introduction
We will delve into the captivating world of Genomic Secondary Analysis, a vital process that transforms raw genomic data into insightful information. This is achieved by aligning the data with a reference genome and identifying variants.
Our journey will take us through the expansive infrastructure of Amazon Web Services (AWS). Here, we'll work with sequenced reference and patient genomes stored as FASTQ GZ files. These files are uploaded to an AWS S3 Bucket, forming the foundation of our analysis.
Our subsequent step involves creating Genomic Analysis Docker Images. These images are deployed into AWS Elastic Container Registry Repositories. From there, we set up an AWS Fargate Compute Environment and an AWS Batch Queue. These components orchestrate the operation of our Docker Images on AWS Elastic Container Service.
As we advance, we'll create AWS Batch Job Definitions for each of our three genomic analysis tools. We'll then establish AWS Step Functions for a State Machine, which runs an Orchestration Workflow. Our ultimate aim is to produce Variant Call Format (VCF) files. These files play a crucial role in analyzing and pinpointing actionable mutations, paving the way for targeted therapies designed to inhibit these specific mutations.
We live in a time of extraordinary progress in Healthcare and Life Sciences, where the prospect of creating precise, patient-specific treatments is not only possible but within our grasp.
Section 2 - Genomics Sequencing Overview
The fascinating journey of genomic sequencing commences with the harvesting of bacterial cells. DNA is carefully extracted from these cells and then broken down into smaller fragments. These fragments are amplified using a technique known as Polymerase Chain Reaction or PCR. A sequencer subsequently deciphers the nucleotide combinations present in these fragments. The end product is a collection of fastq.gz files packed with sequenced genomic data primed for comprehensive analysis.
Bacterial Cells from an Agar Plate are Chemically Treated to Split Open and Release DNA for Purification
DNA is Fragmented into Known Lengths Either Mechanically or Using Enzymatic 'Molecular Scissors'
Polymerase Chain Reaction (PCR) is Utilized to Multiply DNA Fragments, Creating a DNA Library Molecular Scissors'
The DNA Library is Loaded into a Sequencer, Where each 'DNA Read' Identifies the Nucleotide Combinations (A, T, C, and G) in each DNA Fragment
The Sequencer Generates Millions of DNA Reads, Which are Arranged in the Correct Order Using Specialized Software, Like Puzzle Pieces - Once Completed, the Lengthy Genome Sequences are Output as fastq.gz Files Ready for Further Analysis

Genomics Sequencing Overview (Copyright: Adam Jones)
Section 3 - Upload fastq.gz Sequence Files Into AWS S3 Bucket
Following sequencing, we move the sequenced genomic data into an AWS S3 bucket. The FASTQ files that we upload symbolize our DNA sequences.

Uploading Sequence Files Illustration (Copyright: Adam Jones)
Section 4 - Creating BWAMem, SAMTools, and BCFTools Docker Images and Upload to AWS S3 Bucket
Next, we will build Dockerfiles for our essential tools - BWAMem, SAMTools, and BCFTools. But before that, let's delve into what these tools accomplish.
BWAMem is a software package geared towards mapping low-divergent sequences against a substantial reference genome. It excels in aligning sequenced reads of varying lengths, making it a crucial asset in our genomic analysis.
SAMTools offers a collection of utilities for engaging with and processing short DNA sequence read alignments in the SAM, BAM, and CRAM formats. It's an indispensable tool for manipulating alignments, including sorting, merging, indexing, and producing alignments in a per-position format.
BCFTools is purpose-built for variant calling and working with VCFs and BCFs. It is instrumental in managing files that hold information about genetic variants found in a genome.
Armed with a clearer understanding of our tools, we'll proceed to construct Docker Images from these Docker files. These images will subsequently be uploaded to the AWS S3 Bucket, paving the way for our upcoming steps in genomic analysis.
Create Dockerfiles for BWAMem, SAMTools and BCFTools
Build Docker Images for BWAMem, SAMTools, and BCFTools
Upload BWAMem, SAMTools, and BCFTools Docker Images to AWS S3 Bucket

Creating & Uploading Docker Images Illustration (Copyright: Adam Jones)
Section 5 - Pushing BWAMem, SAMTools, and BCFTools Docker Images to AWS ECR Repositories
We will utilize command-line utilities to dispatch the Docker images to AWS Elastic Container Registry repositories. It's important to highlight that each tool within the AWS ECR repository possesses a unique Uniform Resource Identifier or URI. This URI becomes crucial as it will be cited in the JSON File during the ensuing creation step of the AWS Step Functions State Machine.

Pushing Docker Images to AWS ECR Illustration (Copyright: Adam Jones)
Section 6 - Creating the AWS Fargate Compute Environment
We will initiate the AWS Fargate Compute Environment, our central control center for managing and distributing computational resources. AWS Fargate is highly valuable due to its capability to run containers directly, bypassing the need to manage the underlying EC2 instances. Selecting the appropriate Virtual Private Cloud or VPC, subnets, and security group is critical in this procedure. It's also worth noting the pivotal roles played by Fargate Spot Capacity and Virtual CPUs, which are crucial to optimizing our resource utilization.
Initiate Creation of AWS Fargate Compute Environment
Select Enable Fargate Spot Capacity and Maximum vCPUs
Select or Create the Required VPC
Select or Create the Required Subnets
Select or Create the Required Security Group

AWS Fargate Setup Illustration (Copyright: Adam Jones)
Section 7 - Creating the AWS Batch Queue Utilizing AWS Fargate Compute Environment
Setting up the AWS Batch Queue is our next move. This plays a pivotal role in orchestrating job scheduling. The queue's Amazon Resource Name or ARN will be required during the subsequent AWS Step Function State Machine Workflow Start-Execution and Output to AWS S3 step.
Create the AWS Batch Queue - Jobs Will Stay in Queue to be Scheduled to Run in Compute Environment
Orchestration Type for the Queue Will Be AWS Fargate
Select Job Queue Priority and the Fargate Compute Environment (From Previous Step)
This AWS Batch Queue Will Utilize AWS EC2 Spot Instances
Copy ARN (Amazon Resource Name) for the Queue

AWS Batch Queue Creation Illustration (Copyright: Adam Jones)
Section 8 - Creating the AWS Batch Job Definitions
We set up AWS Batch Job Definitions for BWAMem, SAMTools, and BCFTools. This stage involves choosing the orchestration type, configuring the job role, determining the vCPU count, setting the memory capacity, and adding a volume for the Docker Daemon.
Create the 3 AWS Batch Job Definitions (BWAMem, SAMTools, and BCFTools)
Select AWS Fargate for the Orchestration Type
Enter the URI (Uniform Resource Identifier) from the Previous Docker Images to AWS ECR Repositories Step
Specify Job Role Configuration, vCPU Count, and Memory Amount
Specify Mount Points Configuration, Volume Addition for Docker Daemon and Logging Configuration
3 AWS Batch Job Definitions (BWAMem, SAMTools, and BCFTools)

AWS Batch Job Definitions Illustration (Copyright: Adam Jones)
Section 9 - Creating the AWS Step Functions State Machine
Setting up the AWS Step Functions State Machine entails importing the definition JSON, which incorporates the Uniform Resource Identifier or URI that we gathered during the previous step of Pushing BWAMem, SAMTools, and BCFTools Docker Images to AWS ECR Repositories. The design of the State Machine is then displayed, prompting us to input the State Machine name and choose or establish the permissions execution role.
Initiate Creation of AWS Step Functions State Machine
Select a Blank Template
Import Definition JSON - This Definition Will Include the URIs (Uniform Resource Identifier) for Each of the 3 Docker Images (BWAMem, SAMTools, and BCFTools) in the AWS Elastic Container Registry Repositories
The State Machine Design from the JSON Will Be Visible
Enter the State Machine Name, and Select or Create the Permissions Execution Role

AWS Step Functions Setup Illustration (Copyright: Adam Jones)
Section 10 - AWS Step Functions State Machine Workflow
Our workflow commences with the preparation of resources and inputs, employing AWS Fargate to load Docker images. We then apply the BWA-MEM algorithm to map sequenced reads to our reference genome, generating a SAM file. This file is subsequently converted into a sorted, indexed BAM format via SAMtools, facilitating quicker data access. We call variants per chromosome to boost manageability and then employ BCFtools to examine aligned reads coverage and call variants on the reference genome, culminating in a VCF file of detected variants.
Prepares Resources and Inputs for the Workflow, Loading the Tools Docker Images through the AWS Fargate Compute Environment
Uses the BWA-MEM Algorithm for Aligning Sequenced Reads to the Reference Genome, Producing a SAM File
Utilizes SAMtools to Convert the SAM File into a Sorted and Indexed BAM Format for Quicker Access
Calls Variants per Chromosome, Enhancing Manageability and Efficiency
Uses BCFtools to Overview Aligned Reads Coverage and Call Variants on the Reference Genome, Yielding a VCF File of Identified Variants

State Machine Workflow Illustration (Copyright: Adam Jones)
Section 11 - AWS Step Functions State Machine Workflow Start-Execution and Output to AWS S3
Finally, we initiate the execution of the AWS Step Function State Machine Batch Workflow. This step encompasses importing a JSON file that includes the Job Queue Amazon Resource Name or ARN from the previous step of Creating the AWS Batch Queue, the AWS S3 source and output folders, as well as the Chromosomes targeted for analysis. Upon completion of the workflow, the Chromosome VCF output files are stored in the specified AWS S3 folder.
Start AWS Step Function State Machine Batch Workflow
Import JSON Which Will Include the Job Queue ARN (Amazon Resource Name), AWS S3 Source Folder (For fastq.gz Files), AWS S3 Output Folder (For Variant Call Format Files), and Chromosomes for Analysis (2, 3, 7, 8, 9, 10, and 17)
Start Execution of AWS Step Function State Machine Batch Workflow
End-Completion of Workflow
Chromosome VCF Output Files in AWS S3 Folders

Workflow Execution & Output Illustration (Copyright: Adam Jones)
Section 12 - Leveraging Genomic Data for Precision Treatment of Pediatric Medulloblastoma Example
The final Chromosome VCF output files, particularly chromosomes 2, 3, 7, 8, 9, 10, and 17, offer invaluable data for devising a precision pediatric Medulloblastoma treatment plan. These files hold variant calls that record the distinct mutations present in the cancer cells. By carefully analyzing these variant calls, we can pinpoint actionable mutations fueling the Medulloblastoma. Targeted therapies can be utilized to inhibit these mutations based on the actionable mutations identified. This personalized treatment approach enables us to manage Medulloblastoma more effectively and with fewer side effects. Moreover, regularly monitoring these VCF files can steer decisions regarding potential therapy modifications, laying the groundwork for dynamic, adaptable treatment plans.
The End Chromosome VCF Output Files Provide Significant Data for Creating a Precision Pediatric Medulloblastoma Treatment Plan
With the VCF Output Files, the Variant Calls are Further Analyzed to Identify Actionable Mutations that are Driving the Medulloblastoma
Based on the Identified Actionable Mutations, Targeted Therapies that Specifically Inhibit These Mutations Can Be Selected in Combination with the Traditional Surgery, Chemotherapy, and Radiation Therapy (If Applicable) Approaches

Pediatric Medulloblastoma Case Study Illustration (Copyright: Adam Jones)
Section 13 - A Simplified Path with AWS HealthOmics
In conclusion, let's look at the AWS HealthOmics Data Platform. This platform offers a streamlined and more efficient workflow for our data analysis. It eradicates several manual steps from previous workflows, such as creating Docker images, building AWS Fargate Compute Environments, designing AWS Batch Queue, and manually creating an AWS Step Functions State Machine.
In place of these labor-intensive tasks, AWS HealthOmics automates or abstracts these processes, enabling researchers to concentrate on their core competency - data analysis. We'll assume the AWS CloudFormation Template has already been deployed for this discussion. A forthcoming presentation will provide a detailed exploration of the entire AWS HealthOmics Workflow.
Here's a quick snapshot of the AWS HealthOmics Workflow:
Initially, users upload raw sequence data, specifically FASTQ files, to a pre-determined AWS S3 Bucket set up for inputs.
Subsequently, an AWS Lambda Function, activated by an AWS S3 Event Notification associated with this input bucket, scrutinizes file names and triggers the AWS Step Functions Workflow when it detects a pair of FASTQs with matching sample names.
Next, the AWS Step Functions Workflow activates AWS Lambda functions. These functions import the FASTQs into a HealthOmics Sequence Store and commence a pre-configured HealthOmics Workflow for secondary analysis.
Finally, upon the workflow's completion, the VCF Output Files are directed into AWS Lake Formation.
Stay tuned for my upcoming content, where I'll delve into each step of AWS HealthOmics in detail, offering you a comprehensive insight into this pioneering workflow.
Users Transfer Raw Sequence Data, in the form of FASTQ files, to the Pre-Designated AWS S3 Bucket Intended for Inputs
An AWS Lambda Function is Set to be Activated by an AWS S3 Event Notification Linked to the Input AWS S3 Bucket Which Examines the File Names and Sets Off the AWS Step Functions Workflow When it Identifies a Pair of FASTQs with Identical Sample Names
The AWS Step Functions Workflow Triggers AWS Lambda functions to Import FASTQs into a HealthOmics Sequence Store and Initiate a Pre-Set GATK-based Omics Workflow for Secondary Analysis
Upon Workflow Completion, the VCF Output Files Flow Into AWS Lake Formation
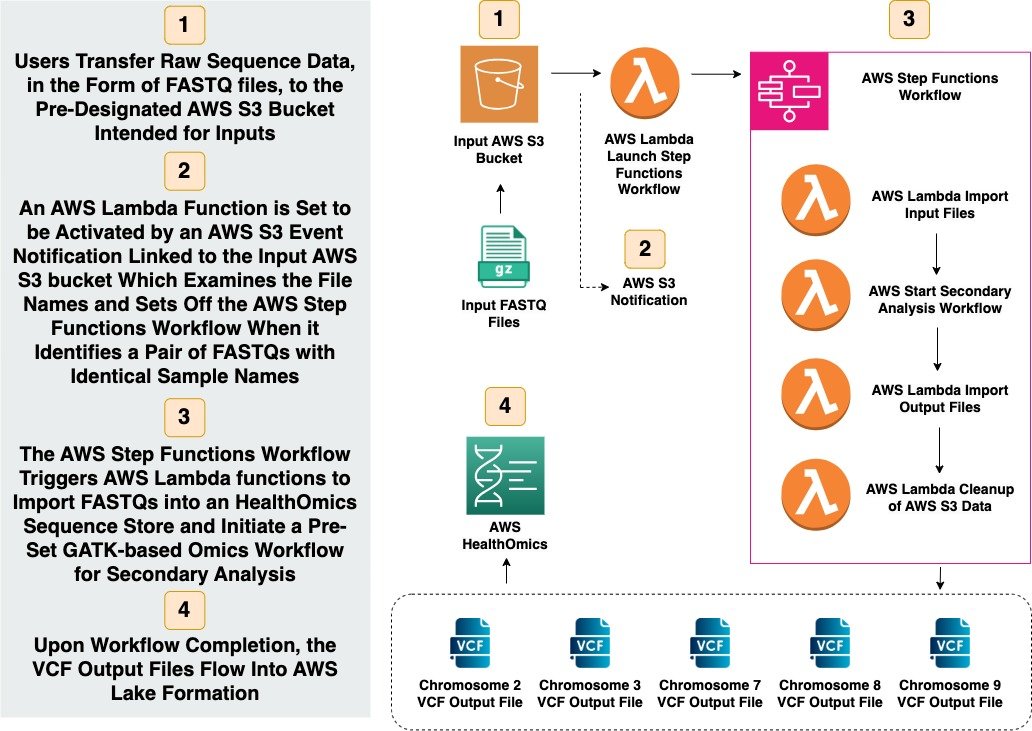
A Simplified Path with AWS HealthOmics Illustration (Copyright: Adam Jones)